Technology and market structure
A major focus of this monograph is the relationship between technology and market structure. High-technology industries are subject to the same market forces as every other industry. However, there are some forces that are particularly important in high-tech, and it is these forces that will be our primary concern. These forces are not ``new.'' Indeed, the forces at work in network industries in 1990s are very similar to those that confronted the telephone and wireless industries in the 1890s.
But forces that were relatively minor in the industrial economy turn out to be critical in the information economy. Second-order effects for industrial goods are often first-order effects for information goods.
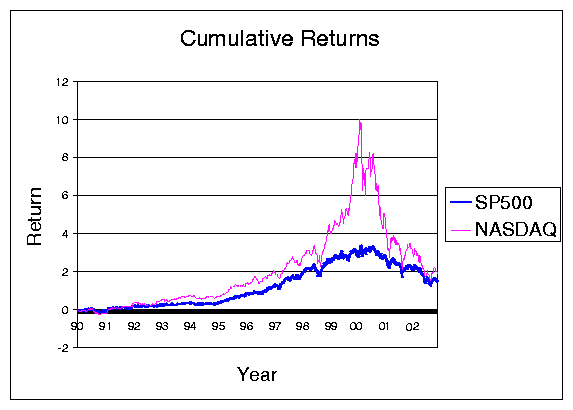 Figure 1: Return on the NASDAQ and S&P 500 during the 1990s.
Figure 1: Return on the NASDAQ and S&P 500 during the 1990s.
Take, for example, cost structures. Constant fixed costs and zero marginal costs are common assumptions for textbook analysis, but are rarely observed for physical products since there are capacity constraints in nearly every production process. But for information goods, this sort of cost structure is very common-indeed it is the baseline case. This is true not just for pure information goods, but even for physical goods like chips. A chip fabrication plant can cost several billion dollars to construct and outfit; but producing an incremental chip only costs a few dollars. It is rare to find cost structures this extreme outside of technology and information industries.
The effects I will discuss involve pricing, switching costs, scale economies, transactions costs, system coordination, and contracting. Each of these topics has been extensively studied in the economics literature. I do not pretend to offer a complete survey of the relevant literature, but will focus on relatively recent material in order to present a snapshot of the state of the art of research in these areas.
I try to refer to particularly significant contributions and other more comprehensive surveys. The intent is to provide an overview of the issues for an economically literate, but non-specialist, audience.
For a step up in technical complexity, I can recommend the survey of network industries in the
Journal of Economic Literature consisting of articles by
Katz and Shapiro [1994],
Besen and Farrell [1994],
Leibowitz and Margolis [1990], and the books by
Shy [2001] and
Vulkan [2003].
Farrell and Klemperer [2003] contains a detailed survey of work involving switching costs and network effects with an extensive bibliography.
For a step down in technical complexity, but with much more emphasis on business strategy, I can recommend
Shapiro and Varian [1998a], which contains many real-world examples.
Intellectual property
There is one major omission from this survey, and that is the role of intellectual property.
When speaking of information and technology used to manipulate information, intellectual property is a critical concern. Copyright law defines the property rights of the product being sold. Patent law defines the conditions that affect the incentives for, and constraints on, innovation in physical devices and, increasingly in software and business processes.
My excuse for the omission of intellectual property from this survey is that this topic is ably covered by my coauthor,
David [2002]. In addition to this piece, I can refer the reader to the surveys by
Gallini and Scotchmer [2001],
Gallini [2002] and
Menell [2000], and the reviews by
Shapiro [2000],
Shapiro [2001].
Samuelson and Varian [2002] describe some recent developments in intellectual property policy.
The Internet boom
First we must confront the question of what happened during the late 1990s. Viewed from 2003, such an exercise is undoubtedly premature, and must be regarded as somewhat speculative. No doubt a clearer view will emerge as we gain more perspective on the period. But at least I will offer one approach to understanding what went on.
I interpret the Internet boom of the late 1990s as an instance of what one might call ``combinatorial innovation.''
Every now and then a technology, or set of technologies, comes along that offers a rich set of components that can be combined and recombined to create new products. The arrival of these components then sets off a technology boom as innovators work through the possibilities.
This is, of course, an old idea in economic history.
Schumpeter [1934], p. 66 refers to ``new combinations of productive means.'' More recently,
Weitzman [1998] used the term ``recombinant growth.''
Gilfillan [1935],
Usher [1954],
Kauffman [1995] and many others describe variations on essentially the same idea.
The attempts to develop interchangeable parts during the early nineteenth century is a good example of a technology revolution driven by combinatorial innovation.
3 The standardization of design (at least in principle) of gears, pullies, chains, cams, and other mechanical devices led to the development of the so-called ``American system of manufacture'' which started in the weapons manufacturing plants of New England but eventually led to a thriving industry in domestic appliances.
A century later the development of the gasoline engine led to another wave of combinatorial innovation as it was incorporated into a variety of devices from motorcycles to automobiles to airplanes.
As Schumpeter points out in several of his writings (e.g.,
Shumpeter [2000]), combinatorial innovation is one of the important reasons why inventions appear in waves, or ``clusters,'' as he calls them.
... as soon as the various kinds of social resistance to something that is fundamentally new and untried have been overcome, it is much easier not only to do the same thing again but also to do similar things in different directions, so that a first success will always produce a cluster. (p 142)
Schumpeter emphasizes a ``demand-side'' explanation of cluster of innovation; one might also consider a complementary ``supply-side'' explanation: since innovators are, in many cases, working with the same components, it is not surprising to see simultaneous innovation, with several innovators coming up with essentially the same invention at almost the same time. There are many well-known examples, including the electric light, the airplane, the automobile, and the telephone.
A third explanation for waves of innovation involves the development of complements. When automobiles were first being sold, where did the paved roads and gasoline engines come from? The answer: the roads were initially the result of the prior decade's bicycle boom, and gasoline was often available at the general store to fuel stationary engines used on farms. These complementary products (and others, such as pneumatic tires) were enough to get the nascent technology going; and once the growth in the automobile industry took off it stimulated further demand for roads, gasoline, oil, and other complementary products. This is an example of an ``indirect network effect,'' which I will examine further in section
10.
The steam engine and the electrical engine also ignited rapid periods of combinatorial innovation. In the middle of the twentieth century, the integrated circuit had a huge impact on the electronics industry. Moore's law has driven the development of ever-more-powerful microelectronic devices, revolutionizing both the communications and the computer industry.
The routers that laid the groundwork for the Internet, the servers that dished up information, and the computers that individuals used to access this information were all enabled by the microprocessor.
But all of these technological revolutions took years, or even decades to work themselves out. As
Hounshell [1984] documents, interchangeable parts took over a century to become truly reliable. Gasoline engines took decades to develop. The microelectronics industry took 30 years to reach its current position.
But the Internet revolution took only a few years. Why was it so rapid compared to the others? One hypothesis is that the Internet revolution was minor compared to the great technological developments of the past. (See, for example,
Gordon [2000].) This may yet prove to be true-it's hard to tell at this point.
But another explanation is that the component parts of the Internet revolution were quite different from the mechanical or electrical devices that drove previous periods of combinatorial growth.
The components of the Internet revolution were not physical devices as all. Instead they were ``just bits.'' They were ideas, standards specifications, protocols, programming languages, and software.
For such immaterial components there were no delays to manufacturer, or shipping costs, or inventory problems,. Unlike gears and pulleys, you can never run out of HTML! A new piece of software could be sent around the world in seconds and innovators everywhere could combine and recombine this software with other components to create a host of new applications.
Web pages, chat rooms, clickable images, web mail, MP3 files, online auctions and exchanges ... the list goes on and on. The important point is that all of these applications were developed from a few basic tools and protocols. They are the result of the combinatorial innovation set off by the Internet, just as the sewing machine was a result of the combinatorial innovation set off by the push for interchangeable parts in the late eighteenth century munitions industry.
Given the lack of physical constraints, it is no wonder that the Internet boom proceeded so rapidly. Indeed, it continues today. As better and more powerful tools have been developed, the pace of innovation have even sped up in some areas, since a broader and broader segment of the population has been able to create online applications easily and quickly.
Twenty years ago the thought that a loosely coupled community of programmers, with no centralized direction or authority, would be able to develop an entire operating system, would have been rejected out of hand. The idea would have been just too absurd. But it has happened: GNU/Linux was not only created online, but has even become respectable and raised a serious threat to very powerful incumbents..
Open source is software is like the primordial soup for combinatorial innovation. All the components are floating around in the broth, bumping up against each other and creating new molecular forms, which themselves become components for future development.
Unlike closed-source software, open source allows programmers and ``wannabe programmers'' to look inside the black box to see how the applications are assembled. This is a tremendous spur to education and innovation.
It has always been so. Look at
Josephson [1959]'s description of the methods of Thomas Edison:
``As he worked constantly over such machines, certain original insights came to him; by dint of may trials, materials long known to others, constructions long accepted were put together in a different way-and there you had an invention.'' (p. 91)
Open source makes the inner workings of software apparent, allowing future Edisions to build on, improve, and use existing programs-combining them to create something that may be quite new.
One force that undoubtedly led to the very rapid dissemination of the web was the fact that HTML was, by construction, open source. All the early web browsers had a button for ``view source,'' which meant that many innovations in design or functionality could immediately be adopted by imitators-and innovators-around the globe.
Perl, Python, Ruby, and other interpreted languages have the same characteristic. There is no ``binary code'' to hide the design of the original author. This allows subsequent users to add on to programs and systems, improving them and making them more powerful.
Financial speculation
Each of the periods of combinatorial innovation referred to in the previous section was accompanied by financial speculation. New technologies that capture the public imagination inevitably lead to an investment boom: Sewing machines, the telegraph, the railroad, the automobile ... the list could be extended indefinitely.
Perhaps the period that bears the most resemblance to the Internet boom is the so-called ``Euphoria of 1923,'' when it was just becoming apparent that broadcast radio could be the next big thing.
The challenge with broadcast radio, as with the Internet, was how to make money from it.
Wireless World, a hobbyist magazine, even sponsored a contest to determine the best business model for radio. The winner was ``a tax on vacuum tubes'' with radio commercials being one of the more unpopular choices.
4
Broadcast radio, of course, set off its own stock market bubble. When the public gets excited about a new technology, a lot of ``dumb money'' comes into the stock market. Bubbles are a common outcome. It may be true that it's hard to start a bubble with rational investors-but not it's not that hard with real people.
Though billions of dollars were lost during the Internet bubble, a substantial fraction of the investment made during this period still has social value. Much has been made of the miles laid of ``dark fiber.'' But it's just as cheap to lay 128 strands of fiber as a single strand, and the
marginal cost of the ``excess'' investment was likely rather low.
The biggest capital investment during the bubble years was probably in human capital. The rush for financial success led to a whole generation of young adults immersing themselves in technology. Just as it was important for teenagers to know about radio during the 1920s and automobiles in the 1950s, it was important to know about computers during the 1990s. ``Being digital'' (whatever that meant) was clearly cool in the 1990s, just as ``being mechanical'' was cool in the 1940s and 1950s.
This knowledge of, and facility with, computers will have large payoffs in the future. It may well be that part of the surge in productivity observed in the late 1990s came from the human capital invested in facility with spreadsheets and web pages, rather than the physical capital represented by PCs and routers. Since the hardware, the software, and the wetware-the human capital-are inexorably linked, it is almost impossible to subject this hypothesis to an econometric test.
Where are we now?
As we have seen, the confluence of Moore's Law, the Internet, digital awareness, and the financial markets led to a period of rapid innovation. The result was excess capacity in virtually every dimension: compute cycles, bandwidth, and even HTML programmers. Al of these things are still valuable-they're just not the source of profit that investors once thought, or hoped, that they would be.
We are now in a period of consolidation. These assets have been, and will continue to be marked to market, to better reflect their true asset value-their potential for future earnings. This process is painful, to be sure, but not that different in principle from what happened to the automobile market or the radio market in the 1930s. We still drive automobiles and listen to the radio, and it is likely that the web-or its successor-will continue to be used in the decades to come.
The challenge now is to understand how to use the capital investment of the 1990s to improve the way that goods and services are produced. Productivity growth has accelerated during the latter part of the 1990s, and, uncharacteristically, continued to grow during the subsequent slump. Is this due to the the use of information technology? Undoubtedly it played a role, though there will continue to be debates about just how important it has been.
Now we are in the quiet phase of combinatorial innovation: the components have been perfected, the initial inventions have been made, but they have not yet been fully incorporated into organizational work practices.
David [1990] has described how the productivity benefits from the electric motor took decades to reach fruition. The real breakthrough came from miniaturization and the possibility of rearranging the production process. Henry Ford, and the entire managerial team, were down on the factor floor every day fine tuning the flow of parts through the assembly line as they perfected the process of mass production.
The challenge facing us now is to re-engineer the flow of information through the enterprise. And not only within the enterprise-the entire value chain is up for grabs. Michael Dell has shown us how direct, digital communication with the end user can be fed into production planning so as to perfect the process of ``mass customization.''
True, the PC is particularly susceptible to this form of organization, given that it is constructed from a relatively small set of standardized components. But Dell's example has already stimulated innovators in a variety of other industries. There are many other examples of of innovative production enabled by information technology that will arise in the future.
The ``New Economy''
There are those that claim that we need a new economics to understand the new economy of bits. I am skeptical. The old economics-or at least the old principles-work remarkably well. Many of the effects that drive the new information economy were there in the old industrial economy-you just have to know where to look.
Effects that were uncommon in the industrial economy-like network effects, switching costs, and the like-are the norm in the information economy. Recent literature that aims to understand the economics of information technology is firmly grounded in the traditional literature. As with technology itself, the innovation comes not in the basic building blocks, the components of economic analysis, but rather the ways in which they are combined.
Let us turn now to this task of describing these ``combinatorial innovations'' in economic thinking.